
- 首页
- 关于我们
- 标准验厂
◇ - 客户验厂
◇- Disney迪士尼验厂
- 沃尔玛(Wal-Mart)验厂
- 好市多(COSTCO)验厂
- 杰西潘尼(JCPenney)验厂
- 孩之宝(hasbro)验厂
- 塔吉特(Target)验厂
- 亚马逊(Amazon)验厂
- 麦当劳(McDonald)验厂
- 雅芳(AVON)验厂
- 宜家(IKEA)验厂
- 星巴克(Starbucks)验厂
- 可口可乐(COCA COLA)验厂
- 家乐福(Carrefour)验厂
- 百思买(BestBuy)验厂
- 乐购(tesco)验厂
- 凯马特(Kmart)验厂
- 利丰(Li&Fung)验厂
- 阿迪达斯(Adidas)验厂
- 彪马(PUMA)验厂
- Argos验厂
- 迪卡侬(Decathlon)验厂
- 史泰博(Staples)验厂
- 美泰(Mattel)验厂
- 梅西(Macy's)验厂
- 耐克(NIKE)验厂
- 西尔斯(Sears)验厂
- 家得宝(Home Depot)验厂
- 玛莎(Marks Spencer)验厂
- Nordstrom验厂
- 抖音验厂
- Apple苹果验厂
- Zara飒拉验厂
- 科尔士(Kohl's)验厂
- 名创优品(MINISO)验厂
- 欧莱雅(LOREAL)验厂
- 沃尔沃斯(Woolworths)验厂
- 奇堡(TCHIBO)验厂
- 体系认证
◇- ISO9001质量管理体系认证
- ISO14001环境管理体系认证
- ISO45001职业健康安全管理体系认证
- ISO22716化妆品质量管理体系认证
- GMPC认证
- GMP/cGMP认证
- TS16949/IATF16949认证
- GRS全球回收标准认证
- GOTS全球有机纺织品认证
- OCS有机含量标准认证
- RCS回收含量声明标准认证
- Oeko-Tex Standard 100认证
- FSC森林认证
- PEFC森林认证
- BRC认证(英国零售商协会)
- SLCP社会劳工整合项目
- Higg工厂环境模块
- ISO13485医疗器械质量管理体系认证
- ISO27001信息安全管理体系认证
- ISO20000信息技术服务管理体系认证
- ISO22000食品安全管理体系认证
- FSSC22000食品安全体系认证
- HACCP食品生产关键点控制管理体系
- GB/T50430建筑行业质量管理体系
- QC080000有害物质过程管理体系认证
- ISO10012测量管理体系认证
- ISO50001能源管理体系认证
- ISO14064碳排放管理体系认证
- ISO22301业务连续性管理体系认证
- 知识产权管理体系(贯标)
- 十环认证(中国环境标志认证)
- 国军标质量管理体系认证
- ISO28000供应链安全管理体系认证
- PAS2060碳中和认证
- ISO14067碳足迹认证
- ISO55001资产管理体系认证
- GB/T31950诚信管理体系认证
- ISO27701隐私信息管理体系认证
- ISO27017云服务信息安全管理体系
- ISO37001反贿赂管理体系认证
- ISO37301合规管理体系认证
- ISO14067碳标签认证
- ISO10015培训管理体系认证
- ISO10002客户投诉管理体系认证
- 医疗器械
◇ - IT行业
◇ - 可持续认证
- 成功案例
- 联系我们

13798281568

第二类体外诊断试剂注册证许可事项变更
办理条件
(一)申请体外诊断试剂注册证书许可事项变更的申请人,应符合以下条件之一:
1.抗原、抗体等主要材料供应商变更的;
2.检测条件、阳性判断值或者参考区间变更的;
3.注册产品技术要求中所设定的项目、指标、试验方法变更的;
4.包装规格、适用机型变更的;
5.产品储存条件或者产品有效期变更的;
6.增加预期用途,如增加临床适应症、增加临床测定用样本类型的;
7.可能影响产品安全性、有效性的其他变更。
(二)有以下情形之一的,不予申请:
1.产品基本反应原理改变;
2.产品阳性判断值或者参考区间改变,并具有新的临床诊断意义;
3.其他影响产品性能的重大改变。
(三)广东省药品监督管理局发放医疗器械注册证书(体外诊断试剂),证书在有效期内。
办理流程
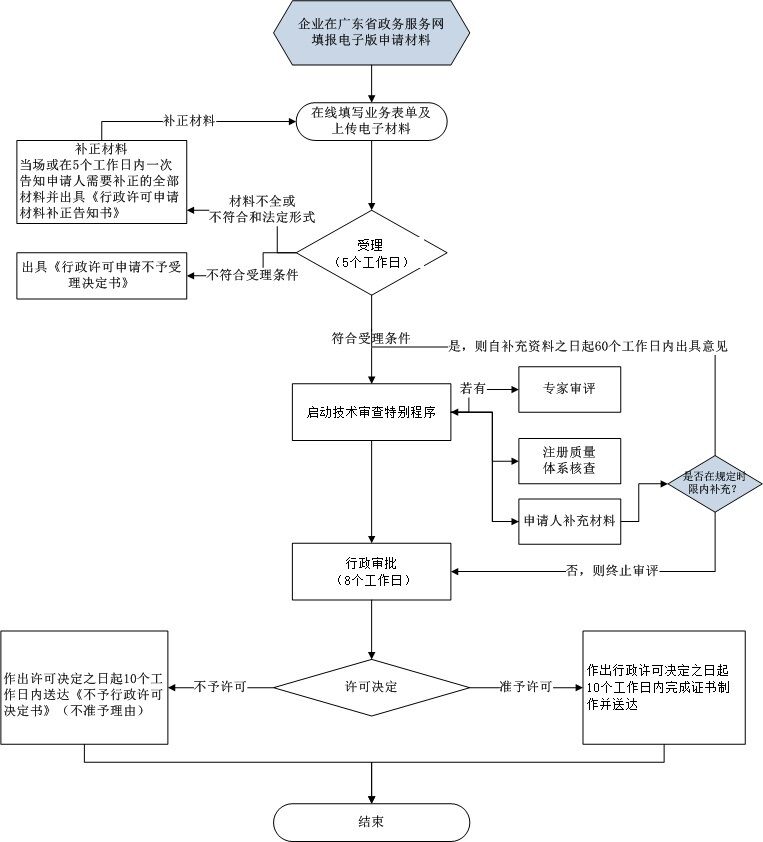
申请材料
(一)医疗器械注册证(体外诊断试剂)许可事项变更申请表
(二)医疗器械注册证(体外诊断试剂)许可事项变更证明性文件
(三)医疗器械注册证(体外诊断试剂)许可事项变更注册人关于变更情况的声明
(四)医疗器械注册证(体外诊断试剂)许可事项变更)原医疗器械注册证及其附件复印件、历次医疗器械注册变更文件复印件
(五)医疗器械注册证(体外诊断试剂)许可事项变更具体变更情况的其他技术资料要求
(六)医疗器械注册证(体外诊断试剂)许可事项变更符合性声明
申请材料形式标准
1、申报资料应有所提交资料目录,每项标题对应的资料应单独编制页码。
2、申报资料应当按目录顺序排列并装订成册。
3、申报资料一式一份,(申请表一式两份,一份与申报资料装订一起,另一份不需要装订),应当使用A4规格纸张打印,内容完整、清楚,不得涂改。凡装订成册的,不得自行拆分。
4、申报资料使用复印件的,复印件应当清晰并与原件一致。申报资料中注明提交原件的,必须按照要求提交原件。
5、各项申报资料中的申请内容应当具有一致性。
6、申报资料均应加盖申请人公章。
7、注册申报资料还需同时提交以下电子文件:(a)申请表,(b)变更部分的对比表,电子文件应为word文档,可编辑、修改。
8、无纸化申报方式的,全部资料通过网上系统提交电子版,不再提交纸质材料。
申报资料的具体要求
(一)申请表
填表说明如下:
1.本表应通过企业网上办事平台(http://219.135.157.143)网上申报端口登陆后在线填报,在线打印并加盖企业公章。
2.申请表封面需加盖企业公章,并加盖骑缝章。申请表“保证书”栏目中的申请人签章,是指法定代表人或负责人签名,加盖企业印章。
3.要求填写的栏目内容原则上使用中文、填写准确、完整、清晰,不得空白、漏填,无相关内容处应填写“∕”。因申请表格式所限而无法填写完整时,请另附附件。
4.无纸化申报的,提交原件扫描版pdf文档,非无纸化申报的,提交原件。
5.前次注册申请情况系指该产品距离本次注册申请最近一次未获准注册的情况。
6.型号、规格应与所提交申请材料实质性内容相对应。
7.分类依据应提供《医疗器械分类目录》的具体品种(对应至品名举例)、或提供分类界定通知文件中的具体条款、或分类界定结果通知书。
8.产品类别及分类编码应根据《医疗器械分类目录》或分类界定意见等相关文件填写。
9.申请人住所栏填写申请人营业执照等相关证明性文件上载明的住所。
10.申请人所在地系指申请人住所所在市。
11.生产地址是指产品实际加工制造的地址。
12.如有其他需要特别加以说明的问题,请在本表“其它需要说明的问题”栏中说明。
(二)证明性文件
境内注册人提供:
1.企业营业执照副本复印件。
2.组织机构代码证复印件。
(三)注册人关于变更情况的声明
1.变更的原因及目的说明。
2.变更可能对产品性能产生影响的技术分析。
3.与产品变化相关的产品风险分析资料。
(四)原医疗器械注册证及其附件复印件、历次医疗器械注册变更文件复印件
(五)具体变更情况的其他技术资料要求
1.变更抗原、抗体等主要材料的供应商,应当提交下列资料:
(1)变更后抗原、抗体等主要材料的研究资料。
(2)分析性能评估资料。
(3)临床试验资料。
(4)变更前、后的产品技术要求、产品说明书。
2.变更检测条件、阳性判断值或参考区间,应当提交下列资料:
(1)变更后的检测条件、阳性判断值或参考区间确定的详细试验资料。
(2)临床试验资料。
(3)变更前、后的产品技术要求、产品说明书。
3.变更产品储存条件和/或有效期,应当提交下列资料:
(1)有关产品稳定性研究的试验资料。
(2)变更前、后的产品技术要求、产品说明书及标签样稿。
4.修改产品技术要求,但不降低产品有效性的变更,应当提交下列资料:
(1)有关分析性能评估的试验资料。
(2)变更前、后的产品技术要求、产品说明书。
5.对产品说明书和/或产品技术要求中文字的修改,但不涉及技术内容的变更,应当提交下列资料:
(1)产品说明书和/或产品技术要求的更改情况说明,说明中应当包含变更情况对比表。
(2)变更前、后的产品说明书和/或产品技术要求。
6.变更包装规格,应当提交下列资料:
(1)变更前、后的产品技术要求、产品说明书和标签样稿(如涉及)。
(2)判断变更的包装规格与已上市包装规格间是否存在性能差异,如存在产品性能差异,需要提交采用变更的包装规格产品进行分析性能评估的试验资料;如产品性能无差异,需要提交变更的包装规格与已上市包装规格之间不存在性能差异的详细说明,具体说明不同包装规格之间的差别及可能产生的影响。
7.变更适用机型,应当提交下列资料:
(1)采用新的适用机型进行分析性能评估的试验资料。
(2)提供变更前、后的产品技术要求、产品说明书和标签样稿(如涉及)。
8.增加临床适应症的变更,应当提交下列资料:
(1)针对增加的临床适应症所进行的分析性能评估资料(如涉及)。
(2)针对增加的临床适应症所进行的临床试验资料。
(3)变更前、后的产品技术要求、产品说明书。
9.增加临床测定用样本类型的变更,应当提交下列资料:
(1)采用增加的临床测定样本类型与已批准的样本类型进行比对的临床试验资料,如增加的样本类型与原批准的样本类型无直接可比性,可以选择与样本类型具可比性的已上市同类产品进行比对的临床试验。
(2)变更前、后的产品技术要求、产品说明书。
10.其他可能影响产品有效性的变更,根据变更情况提供有关变更的试验资料。
11.应当根据产品具体变更情况,提交该变更对产品性能可能产生的影响进行验证的试验资料(如涉及)。
(六)符合性声明
1.注册人声明本产品符合《体外诊断试剂注册管理办法》和相关法规的要求;声明本产品符合现行国家标准、行业标准,并提供符合标准的清单。
2.注册人出具所提交资料真实性的自我保证声明。

本文地址:
版权所有 转载时必须以连接形式注明作者和原始出处
- 上一篇:第二类体外诊断试剂注册证延续
- 下一篇:第二类体外诊断试剂注册证登记事项变更
立即咨询通翔认证验厂专家
13798281568立即在线咨询相关资讯
- 扫一扫咨询






-

-

-

-

-
收起>>
